Cell Biology
Proteins
Proteins have developed a general connotation
in the media and everyday life, especially with nutrition and exercise.
Does the word make you think of a protein shake or a chicken drumstick?
From a cell biologist's point of view, it is the simple idea of putting
amino acid monomers together to form a polymer, a protein macromolecule
containing certain types of information.
Structure
and Function
In biology, structure and function always seem
to be connected in some way. Proteins function by binding to similar
or different proteins. They can also interact with other molecules.
This binding requires shapes that will fit the specified target.
As you can see, the three dimensional structure is very important to protein
function! As a matter of fact, a functioning protein is said to be
in its native conformation. However, the loss of native conformation
(denaturation) can be induced by HEAT, pH, IONIC CONDITIONS, FREEZE THAWING,
AND various CHEMICAL DENATURANTS. As we discussed earlier, the information
in cells are carried in linear sequences. How do 3-D proteins arise?
Experiments
A famous experiment by Christian Anfinsen at the
NIH in 1956 used mercaptoethanol (CH3CH2SH) and urea
to break the dissulfide bonds of ribonuclease A. These enzymes ceased
to function properly so denaturation had occurred. When the two agents
had been removed, the molecules resumed normal enzymic activity and returned
to the original forms! The conclusion was that the linear amino acid
sequence of a polypeptide contained all the directions needed for 3-D structure.
Of course not all proteins fold quickly enough so chaperones bind to them
to speed up the processes.
How do those scientist people figure out the 3-D
structure of proteins? There are different ways to do this, but beware...
the functional state may not be the same as the state a protein is in while
being examined.
One experimental approach is NMR spectroscopy. This will work best
with small proteins.
X-ray diffraction requires a crystalline structure. A thin beam of
X rays of a single wavelength is sent towards a
crystal, and the scattered radiation allows for complex calculations and
analysis. Then they guess randomly or
something :) . Well, I am sure they have the diffraction patterns
and spots of different positions and intensities
figured out so that 3-D structure can be determined.
Primary
Structure
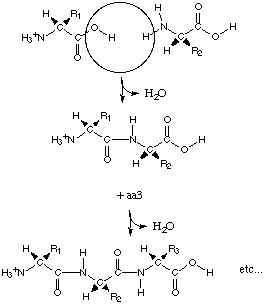
Let's move into the concepts behind the framework
of proteins. The linear sequence of monomers is held together by
peptide bonds. The loss of water (hydrolysis) accompanies the linking
of two monomers. The reason for the interaction between the carboxyl
group and the amino groups is that the new structure will be resonance
stabilized. Note that the peptide bond will not rotate like a single
bond because of its resonance structure which will make it a double bond
that is sp2 hybridized (orgo chem, ugh!). Only the alpha
carbons will rotate.
Secondary
Structure
Here's a gen chem review. Hydrogen
bonds (represented by the striped line in the picture of 2 water molecules)
form between an electronegative atom with a lone pair of electrons (like
oxygen or nitrogen) and a hydrogen (that is bonded to another electronegative
atom). This occurs because the hydrogen has a very partial positive
charge due to its polar covalent bond. The lone pair of electrons
will form a hydrogen bond with the electrophilic proton. This helps
to hold DNA helices together, but they also occur between about every 4
(actually 3.6) residues in an alpha helix protein. Residue is the
term describing amino acids incorporated into a polypeptide chain.
Now here are a few things to note about the alpha
helix.
1) The R groups are projected outwards.
2) The side of an alpha helix facing into a polar
solvent can project only polar residues while the other side can contain
only nonpolar side chains. This type of protein would be amphipathic
(containing both polar and nonpolar characteristics).
3) Stability is increased from the numerous H
bonds parallel to the polymer's long axis.
Tertiary
Structure
The R groups of amino acids become important at
the level of tertiary structure because of interaction between nonadjacent
polypeptides. Relationships between distant monomers, the environment,
and proteins create implications. For example, various amino acids
coming out of a ribosome into aqueous solvent will react differently.
Remember amino acids can be polar charged, polar uncharged, or nonpolar.
In fact a DRIVING FORCE OF PROTEIN FOLDING IS TO INTERNALIZE HYDROPHOBIC
R-GROUPS. A variety of noncovalent interactions like ionic/electrostatic
bonds, hydrogen bonds, Van der Waals forces, and disulfide bonds (between
cysteine sulfhydryl groups) all help proteins to fold to hide these nonpolar
sides. Autonomous folding regions (units) are called domains.
They are parts of a protein that would still look like a full protein if
you cut it out of the whole structure. Examples of DNA binding domains
(or structural motifs) include helix-turn-helix, cysteine-histine zinc
finger, cysteine-cysteine zinc finger, and leucine zipper. A mosaic
is a multiple domain.
Quaternary
Structure
So we've gone from amino acids to polypeptides
to folds of protein (polypeptides) to this. The quaternary structure
of proteins can be determinate or indeterminate (i.e., it is hard to say
where individual units of actin muscle filaments start and stop.)
The subunits are held together by noncovalent interactions (remember the
disulfide bonds?) This creates multiple protein complexes.
Implications
of Protein Binding
Here are some implications of proteins functioning
by binding:
1) 3-D structure is very important. The
specificity of proteins to whatever they are supposed to bind to can be
affected by conformational changes.
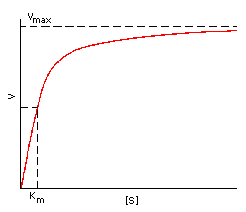 2)
Saturation kinetics compares the rate of dissociation to the rate of association
so that enzyme affinity for a substrate can be expressed by the Michaelis
constant:
2)
Saturation kinetics compares the rate of dissociation to the rate of association
so that enzyme affinity for a substrate can be expressed by the Michaelis
constant:
KM = (K2 + Kcat)/K1
which is approximately K2/K1 since Kcat is negligible
(K2 is rate constant of dissociation, Kcat is progress
of reaction towards product formation, K1 is rate constant of
association, and KM is a ratio of dissociation to association).
The take home message is that a SMALL KM MEANS A HIGHER AFFINITY
FOR THE SUBSTRATE so that the rate would be near its limit at Vmax.
The rate of the enzymatic activity would be proportional to the substrate
concentration.
Some important lessons from all
of this is that conformations are often more conserved than the actual
linear sequence. How do different amino acid sequences specify the
same structure? Many of the R groups are similar enough to where
certain amino acids can take the place of others and still give the same
result in the protein structure. What does this conservation of conformations
also hint at? One idea is that these molecules originated from the
same ancestor. Also these proteins are required for successful creation
of products necessary for survival. The globin family and the TIM
barrel (which includes 30 different proteins, but no functional similarity)
have different amino acid sequences but specify the same structure.
Another point is that binding is often determined
by only a small number of amino acids. For example, millions of immunoglobulins
have the same overall structure, but there are millions of different specific
binding activities. The reason: there are sequence differences in
their 6 small surface loops.
Back
to main page