
|
Antisense oligonucleotides are 6-60 base polymers with sequence
complementary to a selected nucleic acid which hybridize to
(i) mRNA causing
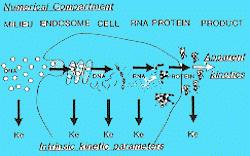
sequence specific inhibition of translation and/or RNAse H degradation; Phosphodiester oligonucleotides are very unstable in blood - t½ is about 5 min. As a result, many backbone modifications have been investigated to increase stability, and most effort has been directed towards nuclease-resistant phosphorothioate (PS) oligonucleotides, i.e., the non-bridging phosphate bound oxygen is replaced by sulfur. "Hybrid" PS-oligonucleotides where a number of 3¢- and 5¢ nucleotides are further modified and "dumbbell", i.e., linked double stranded sequences, have more recently been investigated, because such strategies further stabilize the oligonucleotide. Oligonucleotides (<5000 kDa) are transported across the capillary bed, with the exception of the brain and gonads, and interact with saturable and specific binding sites on cell surfaces, the number of which depends on tissue type; they are also more abundant on dividing cells. Endocytotic internalization occurs with intracellular distribution to endosomes (non-bioavailable) and also to the nucleus (bioavailable). After intravenous administration, initial distribution is very rapid (t½ up to 20 min) with plasma elimination half-lives of 15 to 50 hr or more in most animal species based on radioactivity. Distribution of oligonucleotides generally favors the kidney, liver, lung and spleen, and the elimination half-lives from individual tissues may vary. With more stable oligonucleotides, the majority of the measured plasma/tissue level represents the intact agent; however, backbone degradation does occur (kidney and liver). Both unchanged and degraded oligonucleotide are excreted in urine.
While the oligonucleotide field is still in its infancy, the disposition and pharmacokinetic aspects appear to be mainly analogous to those employed with conventional drugs (Agrawal et. al., 1995; Zhang et. al., 1995). Future progress will probably come in the areas of tissue selective delivery, optimizing intranuclear bioavailability, and understanding of the degradation mechanism(s). |

 |