
|
VIRAL VECTORS IN GENE THERAPY NON-VIRAL VECTORS IN GENE THERAPY
|
VIRAL VECTORS IN GENE THERAPYViral vectors are one of the major vechicles used by scientists in gene therapy to get their sequences expressed in the proper host. There are a myrid of possible viral vectors. This is an area of continual new development in gene therapy.
DEVELOPMENT OF VIRAL VECTORSThe modification of viruses for the delivery of exogenous genes was first reported in 1968. These early attempts using the tobacco mosaic virus showed that viruses could be used to transfer specific genetic material into cells. Studies rapidly shifted to viruses capable of infecting mammalian cells and led to development of a recombinantly modified simian papilloma virus, SV40. With the advent of the now commonplace recombinant DNA techniques, these vectors were capable of transferring and expressing the rabbit -globulin gene in culture. The first recombinant vectors of relevance to the field of human gene therapy were the retroviruses. The development of helper-free packaging cell lines in 1989 marked the advent of retroviral vectors as efficient gene therapeutic agents.
Retroviral and other viral vectorsWhile other recombinant viral vector systems have been developed, retroviral vectors remain the most popular vector system for gene therapy protocols. This may in part be due to their historical significance as the first vectors developed for efficient gene therapy and the infancy of the field of gene therapy. Vector systems to be described in this lecture include retroviral, adenoviral, adeno-associated, herpes simplex, and vaccinia vectors. Discussion will focus on the basic biology of these viruses and vector preparation, as well as their applications and limitations in gene therapeutics.
RETRO-VIRAL VECTORSBASICS OF THE RETROVIRUS VIRION AND INFECTIONRetrovirus virions contain a protein capsid that is lipid encapsulated. Virions range in diameter from 80 to 130 nm. The viral genome is encased within the capsid along with the proteins integrase and reverse transcriptase. The genome consists of two identical positive (sense) single-stranded RNA molecules ranging in size from 3.5 to 10 kilobases. Following cellular entry, the reverse transcriptase synthesizes viral DNA using the viral RNA as its template. The cellular machinery then synthesizes the complementary DNA which is then circularized and inserted into the host genome. Following insertion, the viral genome is transcribed and viral replication is completed. The majority of retroviruses are oncogenic although the degree to which they cause tumors varies from class to class. RETRO-VIRAL RECEPTORSRetroviruses of cats and mice are typically classified by host range. This has led to the use of the following termonology. Ecotropic viruses are viruses which use receptors unique to mice and are only able to replicate within the murine species. Xenotropic viruses uses receptors found on all cells in most species except those of mice. Polytropic and amphotropic viruses use different receptors found in both murine and nonmurine species. THE RETRO-VIRAL GENOMEThe retroviral genome consists of little more than the genes essential for viral replication. The prototype and simplest genome to describe is that of the Moloney murine leukemia virus (MMLV) in contrast to the highly complex genomes of the HTLV and HIV retroviruses. The genome can be divided into three transcriptional units: gag, pol and env. The gag region encodes genes which comprise the capsid proteins; the pol region encodes the reverse transcriptase and integrase proteins; and the env region encodes the proteins needed for receptor recognition and envelope anchoring. An important feature of the retroviral genome is the long terminal repeat (LTR) regions found at each of the gene. The LTR plays an important role in initiating viral DNA synthesis and its integration as well as regulating transcription of the viral genes. VECTOR PREPARATION. Following insertion of the desired gene into in the retroviral DNA vector, and maintainance of the proper packaging cell line, it is now a simple matter to prepare retroviral vectors. The retroviral DNA vector is transferred into the packaging cell line using calcium phosphate mediated transfection, a procedure which we will describe in later lectures. After approximately two days for virion production, the virus is harvested, and this virus is then used to infect a second packaging cell line. Doing this will allow you to produce a virus with a variety of host ranges. The flow chart below is a quick schematic of vector production.
Human immunodeficiency viruses (HIV) are the most recently discovered members of the retrovirus family and have led to the new classification of lentivirus. Like other members of the retroviral family, the HIV genome contains the gag, pol and env genes. In addition, several other nonstructural proteins which serve regulatory functions are contained within its genome. Below is the diagram of the HIV genome considered to be one of the most complex among the retroviruses. Notice that the basic genome units (gag, pol and env) and LTRs predominate the genome.
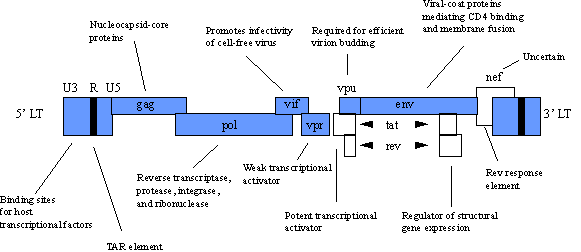
POTENTIAL APPLICATIONS.Rather than an in depth focus on HIV biology, we will discuss the current applications of the HIV virus in gene therapy. HIV-based vectors are recent developments in the field of gene therapy and focus on treatment of AIDS. Vector production is similar to that of MMLV vectors, and HIV vectors possess all the same advantages and disadvantages as their MMLV counterparts. An exciting application of HIV vectors is to use HIV vectors to target genes selectively into HIV-containing cells. The goal is that genes delivered by the HIV vector would allow selective killing of any cell previously or subsequently infected with HIV. An example of how this might be accomplished focuses on the tat and rev genes found in the HIV genome. These genes are also found only in cells infected with HIV. By using a tat and rev induced promoter to drive the expression of a toxin gene, any cell infected with HIV should be selectively killed upon expression of the toxin gene. Of course this system does require that a cell become infected with HIV but once infected the cell would terminate as would the HIV virus. A major concern in using HIV vectors is the fact that there is a strong possibility for genetic recombination between infectious HIV and the HIV vector itself. Such an event would result in the HIV vector acting as an infectious HIV particle. One possible method to overcome this risk would be the inclusion of a suicide gene into the HIV vector genome. The suicide gene would make cells infected with the HIV vector genome sensitive to a drug that would poison only those cells containing the HIV vector genome. An example of a suicide gene is the Herpes simplex virus thymidine kinase gene, and ganciclovir is the drug which would yield selective toxicity. The table below overviews the advantages and disadvantages of retroviral vectors.
ADENOVIRAL VECTORSIntroduction. Adenoviruses were discovered in 1953 as investigators hurriedly attempted to identify the causative agents of the common cold. There are currently 47 distinct serotypes and as many as 93 different particular varieties of adenovirus, all of which generally infect the ocular, respiratory or GI epithelium. In 1977, Frank Graham developed a cell line which enabled the first production of recombinant adenoviruses in a helper free environment. Since this time, adenoviral vectors have recevied much attention as gene transfer agents and currently offer a wide variety of gene therapy applications. Basic structure of the adenovirus virion. Adenoviral virions are icosahedral in shape, 70 to 90 nm in diameter, and are not enveloped. The viral capsid contains 252 protein components the majority of which are three proteins: fiber, penton based and hexon. Fiber and penton base proteins are important in receptor binding and cell internalization, whereas hexon comprises the majority of the viral capsid. The viral genome is large, consisting of a single double-stranded DNA molecule 36 to 38 kilobases in size. Viral DNA replication and transcription are complex, and viral replication and assembly occur only in the nucleus of infected cells. Mature virions are released by cellular disintegration. Adenoviral infection is a highly complex process. It is initiated by the virus binding to the cellular receptor. Internalization occurs via receptor-mediated endocytosis followed by release from the endosome. After endosomal release, the viral capsid undergoes disassembly as it journeys to the nuclear pore. Nuclear entry of the viral DNA is completed upon capsid dissociation, and the viral DNA does not integrate into the host genome but remains in an episomal state.
The adenoviral genome is complex, approximately 36-38 kb long. The E1A and E3 regions are deleted from the majority of adenoviral vectors currently utilized. The E1A region is the first gene to be transcribed upon nuclear entry and is essential for viral replication. The E3 region appears not to be required for viral growth in culture, and removing it allows for a larger DNA insert to be incorporated into the vector. The E3 region has also been shown to play a key role in pathogenesis. Deletion of additional regions of the genome are being studied in the hope of improving the vector for gene therapy applications.
Since adenoviral replication depends on the E1A region of the viral genome, all recombinant adenoviral vectors have this region of its genome deleted, and are referred to as "replication-deficient". Such vectors are capable of infecting a cell only once, no viral propagation occurs, and the infected cell does not die as a result of vector infection. ADENOVIRAL VECTOR PRODUCTIONRecombinant adenoviral vectors are prepared from two components: viral DNA vector and a packaging cell line. The adenoviral DNA vector is a plasmid DNA that contains a portion of the viral genome. It has had the E1A region deleted, and desired genes are cloned into a multicloning site that has been inserted in place of the E1A region of the genome. Unfortunately the large size of the adenoviral genome pervents the use of a single plasmid-based system in vector production. Instead, the adenoviral vector is produced using either in vitro ligation or homologus recombination. Using in vitro ligation, wild-type adenoviral DNA is isolated and cleaved with the restriction endonuclease ClaI. The digested viral DNA is then ligated onto the adenoviral DNA vector containing the gene of interest which has previously been digested with ClaI. The ligated DNA species is then transfected into the packaging cell line. The homologous recombination system can use either ClaI digested viral DNA or a plasmid containing all the adenoviral sequence downstream from the ClaI site. Both the adenoviral DNA vector and the viral DNA component are co-transfected into the packaging cell line. These DNA species are then left to undergo homologous recombination within the cells resulting in vector production. The diagram below illustrates both the in vitro ligation and homologus recombination protocols.
The 293 cell line is utilized for viral production. This is a human kidney cell line which has been stably transfected with the E1A region of the adenoviral genome. This allows the vector to be made and matured within the 293 cell, yet vectors prepared from this cell line will lack the E1A region and remain replication-deficient. As with any viral vector system, one must ensure that the vector produced is free of wild-type contamination. In the adenoviral system, at least two homologous recombination events must occur to obtain wild type virus.
ADVANTAGES & DISADVANTAGESAdenoviral vectors have a high transduction efficiency, are capable of containing DNA inserts up to 8 kilobases, have extremely high viral titers (on the order of 1010-1013), and infect both replicating and differentiated cells. Also, since they lack integration, they can not bring about mutagenic effects caused by random integration into the host genome. Disadvantages of adenoviral vectors include the following: (1) expression is transient since the viral DNA does not integrate into the host, (2) viral proteins are expressed in the adenoviral vector following administration into the host, and (3) adenoviral vectors are an extremely common human pathogen and in vivo delivery may be hampered by prior host immune response to one type virus.
ADENO ASSOCIATED VIRUSESAdeno-associated viruses (AAV) are satellite viruses of other human viruses and require co-infection with either adenovirus or herpes simplex virus (HSV) for their replication. AAV is a parvoviruses. These extremely small icosahedral virions are 18-26 nanometers in diameter and contain a single strand DNA molecule 4-5 kilobases in size. Viruses contain either the sense or antisense strand of the DNA molecule and appear to show no preference for which strand is incorporated into the virion. A unique feature of the DNA molecule contained within the virion is the palindromic sequence at each end, referred to as the inverted terminal repeat (ITR). These ITRs are important in the site-specific integration of AAV DNA into a specific site in chromosome 19. The ability of wild-type AAV to selectively integrate into chromosome 19 made them an attractive candidate for the production of a gene therapy vector that could do the same. Adeno-associated vectors are prepared by replacing the capsid genes with the gene of interest. This has been done using a plasmid-based system analagous to retroviral plasmid vectors but in this case the gene of interest is bracketed by ITRs instead of LTRs. Construction of AAV vectors consists of the recombinant AAV vector plasmid DNA, and a non-rescuable AAV helper plasmid which encodes for the AAV capsid proteins. Also required is either wild-type adenovirus or HSV and cell line for viral propagation. Unlike the previous vector systems described, the cell line need not contain any protion of the AAV genome since all required AAV genome elements will be provided by the two plasmids. Cells are first infected with the wild-type adenovirus or HSV, and then both the recombinant AAV vector plasmid DNA and the non-rescuable AAV helper plasmid are co-transfected into the cells. The cells produce mature recombinant AAV vectors as well as wild-type adenovirus or HSV. The wild-type adenovirus or HSV is removed by either density gradient centrifugation or heat inactivation.
ADVANTAGES AND DISADVANTAGESThese vectors have been designed to produce a gene therapy vector with site-specific integration and the ability to infect multiple cell types. Unfortunately, this has not been the case to date for these vectors. Current research focuses on how to regain the site-specific integration sequences into the recombinant vector. These vectors do offer some advantages over other vector systems which include the lack of initiating an immune response, their stability and ability to infect a variety of dividing and non-dividing cells. Unfortunately, they can not incorporate genes larger than 5 kb and must be closely screened for adenoviral or HSV contamination.
HERPES SIMPLEX VIRUS(HSV)Introduction. Herpes simplex virions are highly structured with an overall diameter of 150 to 200 nm. The genome consists of one double-stranded DNA molecule which is 120 to 200 kilobases in length. The virus itself is transmitted by direct contact and replicates in the skin or mucosal membranes before infecting cells of the nervous system. It exhibits both a lytic and a latent function. The lytic cycle results in viral replication and cell death. The latent function allows for the virus to be maintained in the host for an extremely long period of time. It is hoped that the virus can be modified for gene therapy to produce a vector that exhibits only the latent function for long-term gene maintenance and expression with the specific tendency for cells of the central nervous system.
HSV VECTOR PRODUCTIONRecombinant HSV vectors.Because of the many features of the herpes simplex virus (HSV), the following must be taken into consideration for an effectively engineered HSV vector suitable for gene therapy: (1) the vector must be non-cytotoxic for nerve cells and as well for other cell lines; (2) the vector must be unable to carry out the lytic cycle; (3) the vector can establish latency; and (4) there must be persistent and sufficient levels of the desired gene expression once latency has been obtained. Efforts are currently underway to develop such an HSV vector and are by no means complete at present. Because of the large size of the HSV viral vector, recombinant HSV vectors should be capable of containing an inserted gene of greater than 20 kilobases. HSV vectors are created much like every other vector we have described previously. One simply removes the genes from the virus that are essential for replication and replaces them with the desired genes. Others genes which can be removed are those which are nonessential for replication or growth in tissue culture. This allows for a larger DNA insert to be placed into the vector. Again, any portion of the viral genome essential for replication must be placed in an appropriate packaging cell line so that the replication-deficient HSV vector can be produced. Key regions of the HSV genome which must be removed to produce a replication-deficient HSV vector include the ICP4, ICP0, and ICP27. Please refer to the genome map above. Once the packaging cell line has been prepared and the gene of interest has been cloned into the deleted region of the HSV genome, the resulting vector DNA is transfected into the packaging cell line. Following transfection, HSV vector can be isolated from the cells after 4-7 days in culture. Again, purified HSV vectors should be tested for helper contamination before use.
ADVANTANGES OF RECOMBINANT HSV VECTORSThe HSV vectors currently under study have the following advantages as gene transfer reagents. They allow for a large DNA insert of up to or greater than 20 kilobases; they have an extremely high titer, on the same order of that for adenoviruses; and they have been shown to express transgenes for a long period of time in the central nervous system so long as the lytic cycle does not occur. The major disadvantages for the HSV systems is: (1) they are far from complete and require much additional engineering to be as efficient as hoped, (2) expression does appear to be transient with the current system suggesting either lytic infection or viral protein expression, and (3) there is a relatively low transduction efficiency such that not many target cells show expression of the desired gene.
AMPLICON BASED VECTORSAmplicon-based vectors rely upon the natural occurrence of defective interfering (DI) particles. DI particles arrise during HSV propagation and are infectious agents which lack portions of the HSV genome for replication. It was observed that plasmid DNA could be packaged into DI particles if the plasmid DNA contained HSV packaging signals. Such DI particles were referred to as DI vectors. Amplicon-based or DI vectors rely on two basic components: the amplicon, plasmid DNA which contains HSV packaging signals and regions in which to clone desired genes, and DI particle production. Following insertion of the desired genes into the amplicon, the amplicon is transfected into any cell line which will allow for efficient HSV propagation. No special sequences are required of the cell line since DI vectors are not recombinants but rely on their natural production to arise. Following amplicon transfection the cells are infected with helper HSV virus. The DI vector is then generated along with helper virus during the viral propagation process. The system can be manipluated so that the ratio of DI particles to helper virus is high.
ADVANTAGES OF AMPLICON BASED VECTORSDI vectors are much easier to prepare than their recombinant counterparts. Both systems share many of the same advantages and disadvantages. Unlike recombinant HSV vectors, DI vectors always contain some level of helper virus contamination which has severely restricted their application in gene therapy. Nonetheless, DI vectors remain a good gene transfer system for many applications outside of the field of gene therapy.
VACCINIA VECTORSVaccinia virus has been utilized as a means for immunization against smallpox and a variety of other infectious agents. Vaccinia virus is a member of the pox virus family. They are large brick shaped virions measuring 300-450 by 170-270 nm. They are enveloped and contain an extremely large genome of 130-200 kilobases. This large genome enables large genes to be inserted into vaccinia-based vector. Vaccinia vectors can infect a large variety of cell types, but a primary concern is their safety. As with smallpox vaccination, the adverse reaction rate for administration of vaccinia vectors is 1 in 50,000 doses. Obviously, vaccinia vectors for gene therapy applications are limited to individuals not previously vaccinated against smallpox or are immune compromised. Recombinant vector production is similar to the other vector systems that we had discussed requiring a viral vector DNA and a packaging cell line. These vectors offer the potential to develop a large variety of gene therapy based vaccinations. Currently these applications are not as cost-effective as the more traditional vaccination protocols but this system might allow for the vaccination against diseases presently lacking a vaccine. The system is also being examined in the treatment of HIV, although currently little attention is given to vaccinia-based systems in the field of gene therapy.
NON-VIRAL VECTORS IN GENE THERAPYThe transfer of genetic material using non-viral systems preceded the developed of viral- based vectors. Non-viral vectors, also called physical mechanisms of gene transfer, can be traced back to the work of Avery, MacLeod, and McCarthy, in 1944 which showed that genes were transferred by nucleic acids. Several studies in the early 1960s reported changes in cellular phenotype following exogenous DNA exposure. The first non-viral technique to gain wide acceptance was calcium phosphate-mediated transfection. This system has undergone little change since being well characterized in the early 1970s. It was not until the advent of cationic liposomes in 1988 that non-viral vectors offered an efficient means to transfer genes into cells. Our discussion will divide non-viral vectors into two categories, those which are limited to in vitro applications, and those which have both in vitro and in vivo applications.
Calcium phosphate transfection, initially described in the early 1960s, was refined and systematically improved to result in a standard protocol which has changed little since the early 1970s. In many cases it remains the system of choice for transferring plasmid DNA into a variety of cell cultures and packaging cell lines. It is particularly important in the production of recombinant viral vectors. BASICS OF THE TECHNIQUE. The technique relies on precipitates of plasmid DNA formed by its interaction with calcium ions. It is a very inexpensive and simple technique to perform. Plasmid DNA is mixed in a solution of calcium chloride, and then is added to a phosphate- buffered solution. Over a period of 20 minutes a fine precipitate forms in the solution, and this solution is then added directly to the cells in culture. Transfer efficiency, the number of cells which express the desired gene, is usually quite limited and only reaches levels greater than 10% in a few specific cell lines. In many cases, the level is less than 1%. Transfection efficiencies can be improved in some cell lines by 'shocking' the cells with DMSO or glycerol. Cells can be either transiently or stably transfected using this technique. Levels of stable transfectants can be improved using bis-hydroxyethylaminoethansulfonate (BES). The DNA precipitates are thought to enter the cell by endocytosis. Although this technique has minimal cellular toxicity, and is both simple and inexpensive, the low level of transgene expression prompted development of other techniques. Little or no transgene expression has been observed in vivo following calcium phosphate transfection. DEAE-dextran is another compound that interacts and delivers DNA into cells. It is more reproducible than calcium phosphate transfection but only works in a select few cell lines. Also, DEAE-dextran-mediated gene transfer results only in transient transfections. The exact mechanism of DNA uptake mediated by this technique is not understood. B.MICROINJECTIONMicroinjection delivers plasmid DNA directly into the cell's nucleus. Using the light microscope, a glass pipette is guided into the nucleus and a small amount of DNA or RNA injected. In doing so, both cytoplasmic and lysosomal degradation of the injected material is avoided and efficient gene expression can be expected from the surviving cells. Unfortunately, this technique is extremely labor intensive and requires well isolated cells. Since safety and efficiency can be closely monitored using microinjection, it may one day prove useful in germline gene therapy applications. Microinjection of a particular gene into a cell at a specific time during development has the potential to alter the genotype and phenotype of every cell formed thereafter. Germline modification has already been accomplished in many mammals, which have been termed transgenic animals, ranging in size from mice to cattle. Germline modifications in humans for the correction of specific disorders is far from a present reality and is fraught with ethical considerations.
Electroporation is the application of high voltage to a mixture of DNA and cells in suspension. The cell-DNA suspension is placed between two electrodes and subjected to an electrical pulse. The DNA enters the cells through holes formed in the cellular membrane during the electrical pulse. The DNA is trapped within the cytoplasm upon termination of the electrical pulse. For efficient gene transfer, electroporation depends upon the nature of the electrical pulse, the distance between the electrodes, the ionic strength of the suspension buffer, and the nature of the cells. Best results have often been obtained from rapidly proliferating cells. Electroporation of mammalian cells is an inefficient technique since many cells do not survive the high voltage nature of this procedure. Because of its unique characteristics, electroporation has been difficult to properly design for in vivo applications.
Liposomes were first described in 1965 as a model of cellular membranes and quickly were applied to the delivery of substances to cells. Liposomes entrap DNA by one of two mechanisms which has resulted in their classification as either cationic liposomes or pH-sensitive liposomes. CATIONIC LIPOSOMES are positively charged liposomes which interact with the negatively charged DNA molecules to form a stable complex. Cationic liposomes consist of a positively charged lipid and a co-lipid. Commonly used co-lipids include dioleoyl phosphatidylethanolamine (DOPE) or dioleoyl phosphatidylcholine (DOPC). Co-lipids, also called helper lipids, are in most cases required for stabilization of liposome complex. A variety of positively charged lipid formulations are commercially available and many other are under development. One of the most frequently cited cationic lipids is lipofectin. Lipofectin is a commercially available cationic lipid first reported by Phil Felgner in 1987 to deliver genes to cells in culture. Lipofectin is a mixture of N-[1-(2, 3-dioleyloyx) propyl]-N-N-N-trimethyl ammonia chloride (DOTMA) and DNA and lipofectin interact spontaneously to form complexes that have a 100% loading efficiency. In other words, all of the DNA is complexed with the lipofectin, provided enough lipofectin is available. It is assumed that the negative charge of the DNA molecule interacts with the positively charged groups of the DOTMA. The lipid:DNA ratio and overall lipid concentrations used in forming these complexes are extremely important for efficient gene transfer and vary with application. Lipofectin has been used to deliver linear DNA, plasmid DNA, and RNA to a variety of cells in culture. Shortly after its introduction, it was shown that lipofectin could be used to deliver genes in vivo. Following intravenous administration of lipofectin-DNA complexes, both the lung and liver showed marked affinity for uptake of these complexes and transgene expression. Injection of these complexes into other tissues has had varying results and, for the most part, are much less efficient than lipofectin-mediated gene transfer into either the lung or the liver. pH-SENSITIVE, or NEGATIVELY CHARGED LIPOSOMES, entrap DNA rather than complex with it. Since both the DNA and the lipid are similarly charged, repulsion rather than complex formation occurs. Yet, some DNA does manage to get entrapped within the aqueous interior of these liposomes. In some cases, these liposomes are destabilized by low pH and hence the term pH- sensitive. To date, cationic liposomes have been much more efficient at gene delivery both in vivo and in vitro than pH-sensitive liposomes. pH-sensitive liposomes have the potential to be much more efficient at in vivo DNA delivery than their cationic counterparts and should be able to do so with reduced toxicity and interference from serum protein. APPLICATIONS Liposomes offer several advantages in delivering genes to cells.
In addition, liposomes overcome problems inherent with viral vectors - specifically concerns of immunogenicity and replication competent virus contamination. The ability to chemically synthesize a wide variety of liposomes has resulted in a highly adaptable and flexible system capable of gene delivery both in vitro and in vivo. As liposome technology is better understood, it should be possible to produce reagents with improved in vivo gene delivery into specific tissues. Studies involving protein-DNA-liposome complexes are already showing promise in the ability to target DNA delivery into specific cells. Current limitations regarding in vivo application of liposomes revolve around the low transfection efficiencies and transient gene expression. Also, liposomes display a small degree of cellular toxicity and appear to be inhibited by serum components. The ability to overcome these problems should greatly facilitate their application to a variety of gene delivery mechanisms.
All gene transfer vector systems rely heavily on plasmid DNA. Surprisingly, transgene expression has been observed in rodent and primate muscle following intramuscular injection of plasmid DNA. The simplicity of injecting plasmid DNA into muscle with a syringe has greatly influenced many aspects of gene therapy research. Tissues which exhibit transgene expression following plasmid DNA injection include the thymus, skin, cardiac muscle, and skeletal muscle. Of these tissues, long-term transgene expression was observed only in striated muscle. Expression in mouse skeletal muscle has been observed for greater than 19 months following a single intramuscular injection. It has been assumed that the plasmid DNA must have entered the nucleus in order for gene expression to occur. Plasmid DNA does not undergo changes in its methylation pattern suggesting that it has not replicated following its injection into muscle. Also, the plasmid DNA does not integrate into the host genome following intramuscular injection. A variety of factors influence the effectiveness of gene transfer into muscle by intramuscular plasmid DNA injection. Multiple injections of plasmid DNA improve the overall expression obtained from a single muscle, but following a single injection transgene expression is observed in less than 1% of the total myofibers. A variety of injection solutions have been tested and all those with physiological osmolarity work similarly. Preinjection of sucrose solutions into the muscle slightly improves transgene expression levels. Stimulating the muscle to proliferate prior to the plasmid DNA injection greatly enhances the levels of transgene expression for at least one month following plasmid DNA injection. Age and species also appear to influence the effectiveness of this technique. Younger animals appear to show higher gene transfer efficiencies than older. Primate muscle appears to be less efficient in its ability to uptake DNA following its injection than rodent muscle. It has been postulated that both the age and species related differences are due to variability in the connective tissue barriers present in muscles of different age or species. Transgene expression has also been observed following the implantation of pelleted plasmid DNA into the muscle. Currently, no conclusive evidence has been provided for the exact mechanism by which plasmid DNA uptake occurs in muscle or why it appears to be a predominantly muscle- specific phenomenon. Nuclear entry of the plasmid appears to be regulated by the nuclear pore complex. The greatest advantages of intramuscular plasmid DNA injection are its simplicity and the fact that plasmids ranging in size from 2-19 kilobases have been successfully used to transfer genes to muscle. Its greatest limitations are the low percentage of myofibers which express genes following single injection and its restriction to skin, thymus and striated muscle.
Ballistic DNA Injection, also known as particle bombardment, Microprojectile gene transfer, or the GENE-GUN, was first developed for gene transfer into plants. Since its initial introduction, it has been modified to transfer genes into mammalian cells both in vitro Plasmid DNA encoding the gene of interest is coated onto microbeads, and these particles are then accelerated by motive force to penetrate cell membranes. Specifically, the plasmid DNA is precipitated onto 1-3 micron sized gold or tungsten particles. These particles are then placed onto a carrier sheet which is inserted above a discharge chamber. At discharge, the carrier sheet accelerates toward a retaining screen which stops the carrier sheet, yet allows the particles to continue toward the target surface. The force can be generated by a variety of means; the most common include high-voltage electronic discharge, spark discharge, or helium pressure discharge. APPLICATIONS Ballistic DNA injection has been successfully used to transfer genes to a wide variety of cell lines and also to the epidermis, muscle and liver. In vivo applications have predominantly focused on the liver, skin, muscle or other organs which can easily be exposed surgically. Ballistic DNA injection also offers the capacity to deliver precise DNA dosages. Unfortunately, genes delivered by this method are expressed transiently and there is considerable cell damage occurring at the center of the discharge site. DNA-BASED IMMUNIZATION Currently the most exciting application of intramuscular and ballistic plasmid DNA injection is in DNA-based immunization protocols. DNA-based vaccinations are being developed to prevent infectious diseases and cancer. Cells which express a foreign protein and subsequently present that protein on their surface are more likely to result in a cell-mediated immune response. Such a response may be more important in fighting lateral viral infections such as those caused by HIV, HSV, CMV, or RSV. Techniques which inject a foreign antigen usually only yield an antibody-mediated immunity. Both the gene gun and direct intramuscular plasmid DNA injection are being used in protocols to immunize against HIV, hepatitis B and C, HSV, influenza, papilloma, tuberculosis, RSV, CMV, Lyme disease, Helicobacter pylori, malaria and Mycoplasma pulmonis. Because of their simplicity, intramuscular or ballistic plasmid DNA injection techniques may offer advantages over viral vectors for development of DNA-based immunizations. VECTOR COMPARISION
TARGETED VECTOR SYSTEMSTargeting gene expression to specific cells or tissues is currently an intensive area of gene transfer research. One of the largest hurdles accompanying gene therapy is the inability to target genes or their expression into specific cells at sufficient quantities. Targeting of gene delivery by exploiting specific cellular properties has been referred to as transductional targeting. Transductional targeting involves altering the gene transfer vector so that the vector delivers genes only to specific cells. This can be accomplished by relying upon characteristics of the target cells, such as their proliferation rate or cell-surface receptors. Another means to accomplish targeted gene expression is aimed at the level of transcription. Transcriptional targeting of gene transfer agents focuses on restricting transgene expression in target cells through the use of upstream genetic elements such as cell-specific promoters and/or enhancer elements. Additional details concerning transcriptional targeting strategies will be discussed during the lecture on regulation of gene expression. Gene therapies which ablate specific genes can also be considered a means of targeting gene expression. Ablation strategies are commonly used in cancer gene therapies and the goal is the removal of a specific gene product. Ablation can be targeted at either the DNA, RNA or protein level. Ablation strategies aimed at the DNA level do so by using antisense molecules to form triplex DNA while those aimed at the RNA use either catalytic or noncatalytic antisense agents. Ablation strategies aimed at the protein level do so by forming dominant-negative mutations to inactivate the desired protein or by using intracellular single chain antibodies to entrap the targeted protein. TRANSDUCTIONAL TARGETING Targeting gene vectors based on the rate of cellular division relies more on the characteristics of the target cells than on the gene transfer vector itself. Retroviral vectors serve as a prime example for this particular type of targeting. Retroviral vectors, as you may remember, infect only rapidly dividing cells. Thus, if your target cell population is rapidly dividing, but is surrounded by cells which are not, a retroviral vector would provide a means to transfer the desired gene specifically to those cells. These strategies are very attractive in targeting tumor cells. TARGETING CELL SURFACE MOLECULES Another transductional targeting strategy is aimed at transferring genes to cells expressing specific cell-surface molceules. Attempts have been made to target specific cellular receptors using modified retroviral vectors, liposomes, and molecular conjugates. Retroviral vectors infect cells by the
binding of the viral envelope glycoprotein to receptor molecules expressed on
the surface of the cell. These binding characteristics are responsible for the
ecotropic, xenotropic and amphotropic terminology discussed during the lecture
on retroviral vectors. The fact that natively occurring differences in the
retroviral envelope glycoprotein alters its interaction with specific cells has
led to a variety of approaches aimed at specifically altering the retroviral
envelope glycoproteins to interact only with cellular receptors expressed by the
target cells.
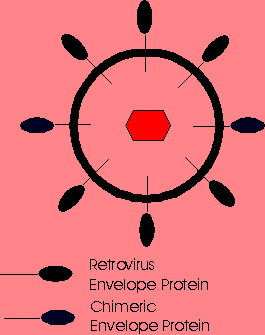 Chemical attachment of lactose to the surface of ecotropic MMLV vectors has permitted specific infection into hepatocytes via the asialoglycoprotein receptor. Thus, chemical modification of the viral surface can be used to target genes to specific cells provided that the ligand can be attached to the virion and that the modifed virion can still transfer genes to the cell. Molecular bridging has also been used to target retroviral vectors to specific cells. Molecular bridging is accomplished by biotinylating two molecules and then using streptavidin as a linker. An antibody specific for the viral envelope glycoprotein is first biotinylated and then mixed with the retroviral vector. The bioltinylated ligand and the streptavidin are then mixed with the vector-antibody complex. Since a single streptavidin can bind 4 biotins, the streptavidin acts as a bridge binding the ligand to the vector at the viral envelope glycoprotein when proper ratios of each are used. Molecular bridging of retroviral vectors with ligands targeting the epidermal growth factor receptor, insulin receptors and the major histocompability complex antigens have transferred genes to the targeted cell populations. Unfortunately, similar strategies using lectins targeting other receptors have not been successful. This would suggest that not all cell surface receptors provide an opportunity for gene transfer. Also, molecularly-bridged vectors, even when successful, resulted in very low levels of infectivity, such that at most, 1 to 2% of the molecularly-bridged virions infected the target cells. FUSION PROTEIN TARGETING Retroviral vectors have also been targeted to specifc cell populations by altering the gene encoding its envelope glycoprotein. This was initially attempted by fusing a gene for a single chain antibody (sFv) to the region of the envelope gene encoding its N- terminal amino acids. The resulting envelope-sFv fusion protein would have the sFv presented outward from the vector and hence available for cell binding while the C-terminal region of the glycoprotein would project into the vector allowing proper virion assembly. This technique has been attempted using many different sFvs and other proteins capable of specifically attaching to cellular receptors. Most have resulted in very low infectivities, less than 3%, and poor virion stability. The most successful targeted retroviral vector reported to date has had the region encoding for the 150 N-terminal amino acids of the envelope protein replaced by sequences encoding for 150 amino acids of erythropoietin (EPO) (3). These vectors were prepared in a packaging cell line so that the final retroviral virion would contain both wild-type ecotropic envelope glycoproteins and EPO-envelope fusion proteins. Such vectors infected only human cells bearing the EPO receptor. These vectors demonstrated good levels of infectivity as well as stability, most likely resulting from the presence of the wild-type envelope protein within the virion. TARGETED LIPOSOMES Liposomes can be prepared to incorporate specific proteins onto their surface. When combined with DNA, such liposomes should have the ability to deliver genes to specific cell populations. Molecules which have been incorporated into the surface of liposomes include monoclonal antibodies, carbohydrate ligands, and protein ligands. This should increase the targeting efficiency of the liposome for the desired cell type. Unfortunately, currently studied systems lack specificity and deliver genes to cells not containing the selected receptor. MOLECULAR CONJUGATE VECTORS A molecular conjugate vector consists of plasmid DNA and an attached ligand. The ligand confers cellular specificity of the conjugate and must also bind to the plasmid DNA. Gene transfer is thought to be accomplished by receptor-mediated endocytosis of the plasmid DNA-ligand conjugate. A variety of molecular conjugates have been prepared to deliver genes to specific cells but these vectors only yield meaningful transgene expression levels when applied in vitro. Following cellular internalization, molecular conjugates appear to remain trapped within the endosome. Transfection efficiencies have been improved by incorporating adenoviruses or other endosome-lysing agents into the molecular conjugate vector composition. Unfortunately, incorporation of such agents has not improved in vivo gene delivery. As such, these vectors currently remain limited to ex vivo gene therapy protocols. |

 |